Building structure
Here we show how to build various structures, including molecule, crystal, surface and nanoparticles.
The Batoms object is used to build structure from scratch, load structure from file or other objects (ASE, Pymatgen).
From scratch
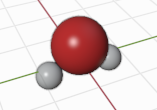
Build a H2O molecule.
from batoms import Batoms
h2o = Batoms(label = "h2o",
species = ["O", "H", "H"],
positions = [[0, 0, 0.40], [0, -0.76, -0.2], [0, 0.76, -0.2]])
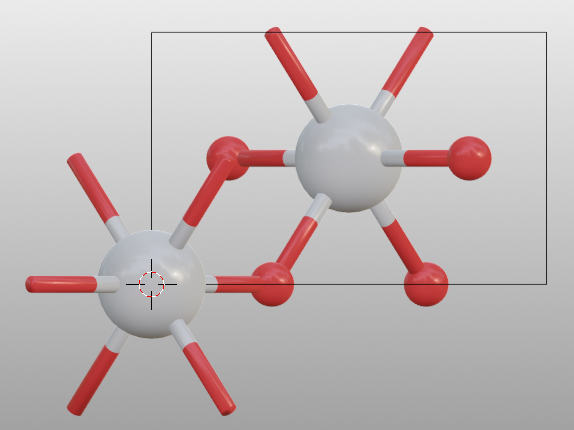
Here is how you could define an gold crystal structure with a lattice constant of 4.08 Å:
from batoms import Batoms
a = 4.08
positions = [[0, 0, 0], [a/2, a/2, 0], [a/2, 0, a/2], [0, a/2, a/2]]
au = Batoms(label = "au",
species = ["Au"]*len(positions),
positions = positions,
pbc = True,
cell = (a, a, a))

Import from file
batoms use ase.io.read function to read file, thus it support various file formats, such as: xyz, pdb, cif, VASP, Espresso, Aims and so on. Please read: https://wiki.fysik.dtu.dk/ase/ase/io/io.html?highlight=read#ase.io.read
from batoms.bio import read
tio2 = read("docs/source/_static/datas/tio2.cif")
ASE
Please read ASE document for building structures using ASE: https://wiki.fysik.dtu.dk/ase/ase/build/build.html?highlight=build#module-ase.build
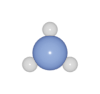
Molecules
ASE defines a number of molecular geometries in the g2 database, which can be load directly.
from ase.build import molecule
from batoms import Batoms
atoms = molecule("NH3")
batoms = Batoms(label = "mol", from_ase = atoms)
batoms.get_image(output = "nh3.png")
The list of available molecules is those from the ase.collections.g2 database:
from ase.collections import g2
g2.names
['PH3', 'P2', 'CH3CHO', 'H2COH', 'CS', 'OCHCHO', 'C3H9C', 'CH3COF',
'CH3CH2OCH3', 'HCOOH', 'HCCl3', 'HOCl', 'H2', 'SH2', 'C2H2',
'C4H4NH', 'CH3SCH3', 'SiH2_s3B1d', 'CH3SH', 'CH3CO', 'CO', 'ClF3',
'SiH4', 'C2H6CHOH', 'CH2NHCH2', 'isobutene', 'HCO', 'bicyclobutane',
'LiF', 'Si', 'C2H6', 'CN', 'ClNO', 'S', 'SiF4', 'H3CNH2',
'methylenecyclopropane', 'CH3CH2OH', 'F', 'NaCl', 'CH3Cl',
'CH3SiH3', 'AlF3', 'C2H3', 'ClF', 'PF3', 'PH2', 'CH3CN',
'cyclobutene', 'CH3ONO', 'SiH3', 'C3H6_D3h', 'CO2', 'NO',
'trans-butane', 'H2CCHCl', 'LiH', 'NH2', 'CH', 'CH2OCH2',
'C6H6', 'CH3CONH2', 'cyclobutane', 'H2CCHCN', 'butadiene', 'C',
'H2CO', 'CH3COOH', 'HCF3', 'CH3S', 'CS2', 'SiH2_s1A1d', 'C4H4S',
'N2H4', 'OH', 'CH3OCH3', 'C5H5N', 'H2O', 'HCl', 'CH2_s1A1d',
'CH3CH2SH', 'CH3NO2', 'Cl', 'Be', 'BCl3', 'C4H4O', 'Al', 'CH3O',
'CH3OH', 'C3H7Cl', 'isobutane', 'Na', 'CCl4', 'CH3CH2O', 'H2CCHF',
'C3H7', 'CH3', 'O3', 'P', 'C2H4', 'NCCN', 'S2', 'AlCl3', 'SiCl4',
'SiO', 'C3H4_D2d', 'H', 'COF2', '2-butyne', 'C2H5', 'BF3', 'N2O',
'F2O', 'SO2', 'H2CCl2', 'CF3CN', 'HCN', 'C2H6NH', 'OCS', 'B', 'ClO',
'C3H8', 'HF', 'O2', 'SO', 'NH', 'C2F4', 'NF3', 'CH2_s3B1d', 'CH3CH2Cl',
'CH3COCl', 'NH3', 'C3H9N', 'CF4', 'C3H6_Cs', 'Si2H6', 'HCOOCH3', 'O',
'CCH', 'N', 'Si2', 'C2H6SO', 'C5H8', 'H2CF2', 'Li2', 'CH2SCH2', 'C2Cl4',
'C3H4_C3v', 'CH3COCH3', 'F2', 'CH4', 'SH', 'H2CCO', 'CH3CH2NH2', 'Li',
'N2', 'Cl2', 'H2O2', 'Na2', 'BeH', 'C3H4_C2v', 'NO2']

PubChem database
More complicated molecules may be obtained using the PubChem API integration. Here is a example of loading tetrabutylammonium bromide structure from PubChem website by search the name of the molecule. https://pubchem.ncbi.nlm.nih.gov/compound/Tetrabutylammonium-bromide.
from batoms.plugins.pubchem import pubchem_search
ssl._create_default_https_context = ssl._create_unverified_context
tbab = pubchem_search(name = "tetrabutylazanium")
batoms = Batoms(label = "mol", from_ase = tbab)
batoms.model_style = 1
batoms.get_image(output = "tbab.png")

Crystal
Create a bulk structure for FCC Au.
from ase.build import bulk
from batoms import Batoms
au = bulk("Au", "fcc", cubic=True)
au = Batoms(label = "au", from_ase = au)
au.get_image(viewport = [1, -0.3, 0.1], output = "au.png")
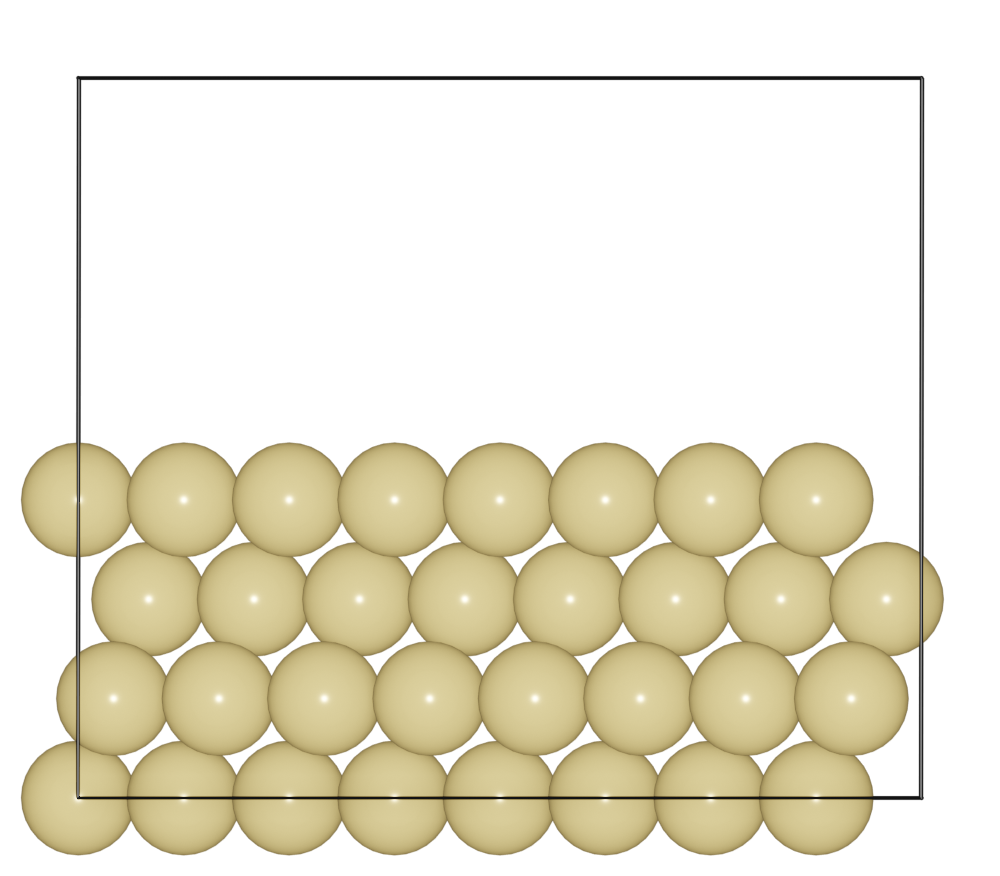
Surface
Create (111) surface for FCC Au.
from ase.build import fcc111
from batoms import Batoms
atoms = fcc111("Au", size = (5, 5, 4), vacuum=0)
au111 = Batoms(label = "au111", from_ase = atoms)
au111.cell[2, 2] += 10
Nanoparticle
Create a nanoparticle using Wulff method:
from ase.cluster import wulff_construction
from batoms import Batoms
surfaces = [(1, 1, 1), (1, 0, 0)]
energies = [1.28, 1.69]
atoms = wulff_construction("Au", surfaces, energies, 500, "fcc")
del atoms[atoms.positions[:, 2] < 0]
nano = Batoms("wulff", from_ase = atoms)
